Resource • Article
Regulatory Guidelines: The Key to Accelerating COVID-19 Studies

The greatest enemy to the containment of coronavirus could be time itself. With close to 35 million cases across the globe, pharmaceutical and biotech companies in the process of developing vaccines and treatments for COVID-19 are fighting across multiple fronts:
- The speed of the disease’s rate of infection
- The immediate need for a viable vaccine to prevent the disease
- The post-trial steps necessary for manufacturing and distribution
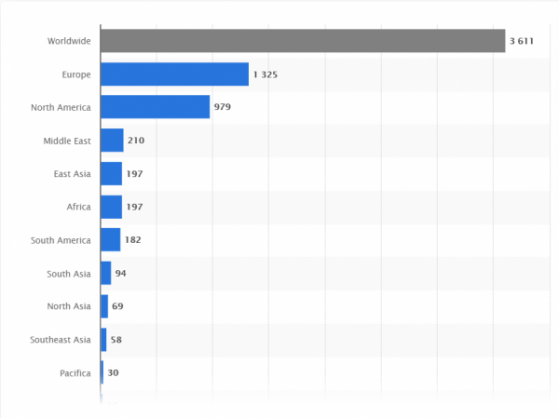
According to the World Health Organization, there are currently 44 vaccines in clinical evaluation, with another 91 at the preclinical stage, stretched across 150+ countries. This, alongside thousands of non-vaccine COVID-19 trials, presents a competitive race across the industry (See Figure 1).
Figure 1: COVID-19 Trial Statistics
Health officials and political leaders may not be on the same page when it comes to the true test of the timeline – when the first COVID-19 vaccine will be made available for widespread usage. However, it is certain that speed is the key to these trials and that regulatory agencies are important stakeholders in moving these studies forward. As the gatekeeper into clinical pathways and the final say in marketing authorization, regulators have the power to accelerate, pause, or stop a product’s development.
Optimizing Regulatory Approval
Consider the standard pathway for moving a drug to market. It can often take from six to seven years to complete all clinical trial phases, receive FDA approval, and facilitate manufacturing and distribution to the general public. The total cost for the journey can exceed $2B.
It is important to note that vaccines often follow a different path than other drugs or medical devices. In fact, the fastest approval for a vaccine was for the mumps in 1977, taking almost four years to complete all of the necessary permissions and licensing before beginning distribution.
At the beginning of 2020, as the COVID-19 pandemic grew more severe, and the need for vaccines and treatments became more critical, the FDA and other global regulatory agencies determined that an option to fast-track potential interventions was needed immediately. In response, fast-track pathways, such as the FDA’s Coronavirus Treatment Acceleration Program (CTAP) program, were initiated. CTAP is a special emergency program for potential coronavirus therapies designed to “move new treatments to patients as quickly as possible, while at the same time finding out whether they are helpful or harmful.”
In the U.S., CTAP includes guidance for:
- An estimated 24-hour turnaround time for study protocol reviews
- An estimated three-hour turnaround time for expanded access requests
- Priority product development and manufacturing support for interventions
- The increase and extension of regulatory staff resources
- A requirement of a 50% efficacy endpoint estimates in clinical trial results
A Better Understanding of Regulatory Guidelines
Before addressing the above CTAP guidelines for accelerated response to COVID-19, it is important to note that the origins of CTAP began in April 2020. The FDA blog post, “A Path Forward,” outlined the initiative to streamline the review and advisement process. In the five months since this post, pharmaceutical companies around the world have jumped into the healthcare market to provide vaccine candidates and other interventions to combat the pandemic, taking advantage of the FDA’s efforts to move studies forward more quickly.
The current status of CTAP, at the end of July 2020, shows the FDA supporting the initiation of 270+ clinical trials, with 570+ additional investigational programs currently under review. Other regulators around the world have done the same with fast-track COVID-19 programs in the European Union (EMA), Russia (Roszdravnadzor), and China (NMPA). The number of companies in the market continues to grow.
How has this affected the FDA? It is important to keep in mind that:
- The CTAP measures are only guidelines, not defined determinations. The FDA is most focused on maintaining the key ingredients of science and data to assess vaccine viability as well as other interventions.
- With potential innovations leveraged by vaccine developers in order to combat COVID-19, the FDA does not want to impede or impair the process by specifying requirements beyond current regulations.
- The core competencies of ensuring patient safety and drug efficacy, along with other variables ranging from patient recruitment to availability of resources, continue to play a role in the development and regulatory process.
- Based on the variables above, the response time for protocol study reviews and expanded access requests from the FDA can range from a single day to 10 days or longer.
Additionally, the FDA’s June 2020 specifications, noting that a vaccine should show at least 50% efficacy in clinical trials, have come under scrutiny by the medical and scientific community. Three promising clinical trials in the race for a COVID-19 vaccine are under investigation:
- The University of Oxford and AstraZeneca vaccine trial has been essentially put on hold in the U.S. as of September 30, 2020. While allowed to restart in other regions, the FDA has not re-initiated the trial and is investigating serious illnesses during Phase 3 of the vaccine trial.
- The FDA has also put a partial hold on Inovio Pharmaceuticals Inc.’s COVID-19 trial, promising further study related to the delivery device for the vaccine.
- Moderna is delaying its request for Emergency Use Authorization (EUA) from the FDA seeking further safety data related to its COVID-19 vaccine.
Finding Common Ground in Clinical Trial Regulations
Faced with open-ended FDA guidance and the importance of speed-to-market, developing COVID-19 vaccines and treatments in the pandemic landscape can be truly challenging. Companies moving ahead with vaccine trials, and seeking to accelerate the process, should keep the following in mind:
- Work closely with the FDA when it comes to clinical trial protocols. Companies should define their clinical studies in detail – from the various trial phases to the end result – and engage in a dialogue with the FDA as early as possible.
- Understand the guidelines and stay up to date. Companies should familiarize themselves with the latest regulations from the FDA and work within the framework. The FDA also recommends adaptive design for a more rapid progression through the phases of clinical development.
- Make room for diversity in your vaccine trials. Since the beginning, the COVID-19 virus has been in flux. Where once the older population was the main demographic for clinical testing, now minorities, children, and teens are seeing greater numbers of infected patients. Be aware of the changes and make amendments to clinical protocols as necessary.
Again, the FDA is looking for science-based data, while also ensuring safety and efficacy within these clinical trials. Meeting these requirements with diligence and confidence can potentially give pharmaceutical companies the key to accelerating their trials for patients in need.
Recognizing regulatory guidelines while preparing for study launch is only one part of the execution strategy for COVID-19 trials. Our next article will focus on monitoring solutions for coronavirus trials.
References
- Times of India.com. “Coronavirus: How Long Does It Really Take To Get A Vaccine Ready? We Explain To You The Process.” June 25, 2020.
- S. Food & Drug Administration. “The Path Forward: Coronavirus Treatment Acceleration Program.” April 20, 2020.
- Shah, Anand, et. al. “FDA Initiatives To Accelerate The Development Of COVID-19 Therapeutics.” Health Affairs.org, August 18, 2020.
Click here to learn more in the Biorasi COVID-19 Article Series
